Kavi's BGGN 213 Portfolio

Class work for bioinformatics class
Class 12
Kavi (PID: )
- Background
- Data Import
- Toy analysis example
- DESeq Analysis
- Volcano Plot
- Save our results
- Pathway Analysis
- Adḍ annotation data
- Save my annotated results
Background
Today we will analyze some RNASeq data from Himes et al. on the effectṡof a common steroid (dexamethasone, also called “dex”) on ariway smooth muscle cells (ASMs).
For this analysis we need two main inputs
countData: a table of counts per gene (in rows) across experiments (in columns)colData: metadata about the design of the experiments. The rows here must match the columns incountData
Data Import
counts <- read.csv("airway_scaledcounts.csv", row.names=1)
metadata <- read.csv("airway_metadata.csv")
head(counts)
SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
ENSG00000000003 723 486 904 445 1170
ENSG00000000005 0 0 0 0 0
ENSG00000000419 467 523 616 371 582
ENSG00000000457 347 258 364 237 318
ENSG00000000460 96 81 73 66 118
ENSG00000000938 0 0 1 0 2
SRR1039517 SRR1039520 SRR1039521
ENSG00000000003 1097 806 604
ENSG00000000005 0 0 0
ENSG00000000419 781 417 509
ENSG00000000457 447 330 324
ENSG00000000460 94 102 74
ENSG00000000938 0 0 0
and the metadata:
metadata
id dex celltype geo_id
1 SRR1039508 control N61311 GSM1275862
2 SRR1039509 treated N61311 GSM1275863
3 SRR1039512 control N052611 GSM1275866
4 SRR1039513 treated N052611 GSM1275867
5 SRR1039516 control N080611 GSM1275870
6 SRR1039517 treated N080611 GSM1275871
7 SRR1039520 control N061011 GSM1275874
8 SRR1039521 treated N061011 GSM1275875
Q1. How many “genes” are in this dataset?
There are 38694 genes in the dataset.
Q2. How many experiments (i.e. columns in
countsor rows inmetadata) are there?
If we use counts: 8
If we use metadata: 8
Q3. How many “control” experiments are there in the dataset?
There are 4 “control” experiments.
Toy analysis example
Contains answers to Q3 and Q4.
- Extract the “control” columns from
counts. - Calculate the mean value for each gene in these “control” columns.
3-4. Do the same for the “treated” columns. 5. Compare these mean values for each gene.
Step 1
control.inds <- metadata$dex == "control"
control.counts <- counts[, control.inds]
head(control.counts)
SRR1039508 SRR1039512 SRR1039516 SRR1039520
ENSG00000000003 723 904 1170 806
ENSG00000000005 0 0 0 0
ENSG00000000419 467 616 582 417
ENSG00000000457 347 364 318 330
ENSG00000000460 96 73 118 102
ENSG00000000938 0 1 2 0
Step 2
control.mean <- rowMeans(control.counts)
Steps 3-4
treated.inds <- metadata$dex == "treated"
treated.counts <- counts[, treated.inds]
head(treated.counts)
SRR1039509 SRR1039513 SRR1039517 SRR1039521
ENSG00000000003 486 445 1097 604
ENSG00000000005 0 0 0 0
ENSG00000000419 523 371 781 509
ENSG00000000457 258 237 447 324
ENSG00000000460 81 66 94 74
ENSG00000000938 0 0 0 0
treated.mean <- rowMeans(treated.counts)
For ease of book-keeping we can store these together in one data frame
called meancounts
meancounts <- data.frame(control.mean,treated.mean)
head(meancounts)
control.mean treated.mean
ENSG00000000003 900.75 658.00
ENSG00000000005 0.00 0.00
ENSG00000000419 520.50 546.00
ENSG00000000457 339.75 316.50
ENSG00000000460 97.25 78.75
ENSG00000000938 0.75 0.00
Step 5 > These are answers to Q5 a and b.
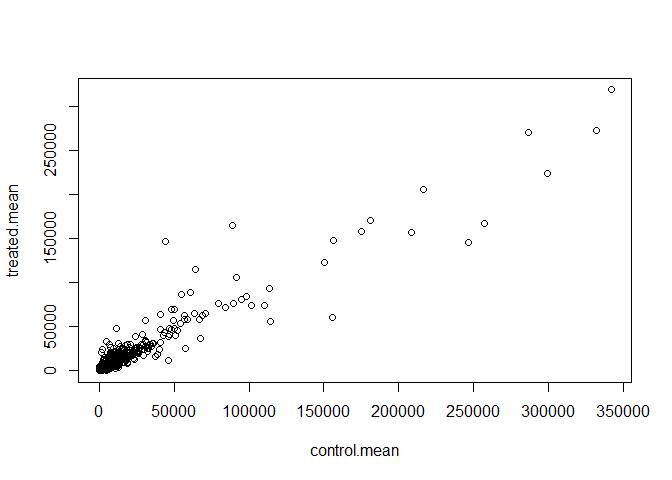
plot(control.mean, treated.mean)
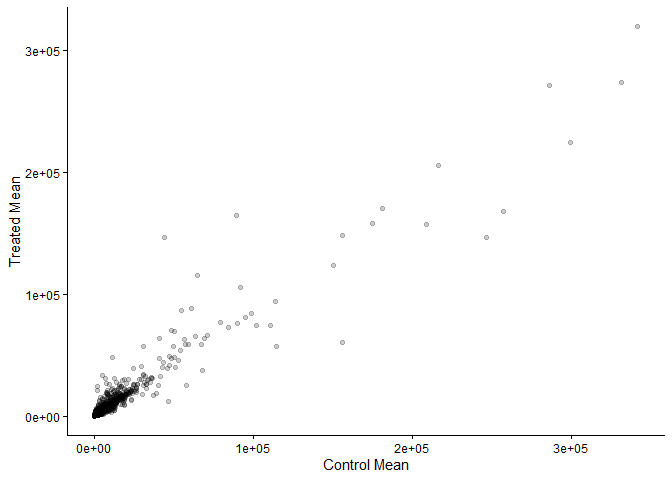
library(ggplot2)
ggplot(meancounts) +
aes(x=control.mean,y=treated.mean) +
labs(x="Control Mean", y="Treated Mean") +
geom_point(alpha=0.2) +
theme_classic()
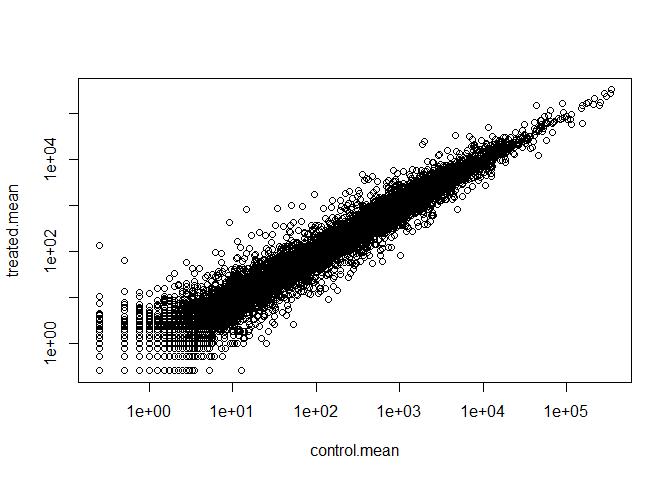
This is the answer to Q6.
plot(control.mean, treated.mean,log="xy")
Warning in xy.coords(x, y, xlabel, ylabel, log): 15032 x values <= 0 omitted
from logarithmic plot
Warning in xy.coords(x, y, xlabel, ylabel, log): 15281 y values <= 0 omitted
from logarithmic plot
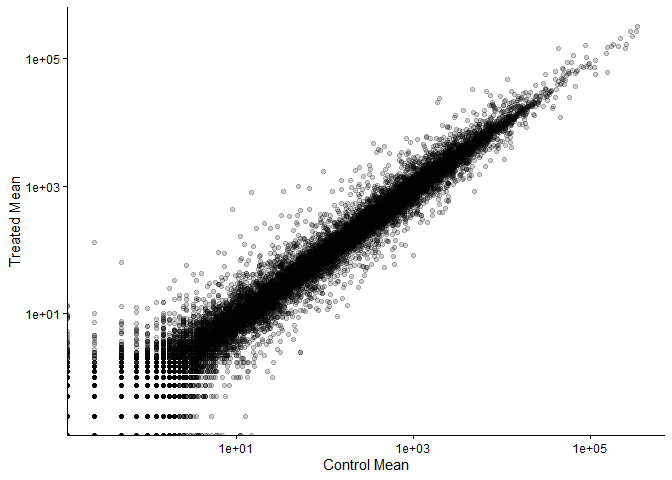
ggplot(meancounts) +
aes(x=control.mean,y=treated.mean) +
labs(x="Control Mean", y="Treated Mean") +
geom_point(alpha=0.2) +
scale_x_log10() +
scale_y_log10() +
theme_classic()
Warning in scale_x_log10(): log-10 transformation introduced infinite values.
Warning in scale_y_log10(): log-10 transformation introduced infinite values.
We use “fold-change” as a way to compare
log2(10/10)
[1] 0
log2(20/10)
[1] 1
log2(10/20)
[1] -1
log2(40/10)
[1] 2
meancounts$log2fc <- log2(meancounts$treated.mean/meancounts$control.mean)
head(meancounts)
control.mean treated.mean log2fc
ENSG00000000003 900.75 658.00 -0.45303916
ENSG00000000005 0.00 0.00 NaN
ENSG00000000419 520.50 546.00 0.06900279
ENSG00000000457 339.75 316.50 -0.10226805
ENSG00000000460 97.25 78.75 -0.30441833
ENSG00000000938 0.75 0.00 -Inf
zero.vals <- which(meancounts[,1:2]==0, arr.ind=TRUE)
to.rm <- unique(zero.vals[,1])
mycounts <- meancounts[-to.rm,]
head(mycounts)
control.mean treated.mean log2fc
ENSG00000000003 900.75 658.00 -0.45303916
ENSG00000000419 520.50 546.00 0.06900279
ENSG00000000457 339.75 316.50 -0.10226805
ENSG00000000460 97.25 78.75 -0.30441833
ENSG00000000971 5219.00 6687.50 0.35769358
ENSG00000001036 2327.00 1785.75 -0.38194109
A common “rule of thumb” threshold for calling something “up” regulated is a log2-fold-change of +2 or greater. For “down” regulated, a log2-fold-change of -2 or lower.
Q8 How many genes are “up” regulated at the +2 log2FC threshold?
nonzero.inds <- rowSums(counts) != 0
mycounts <- meancounts[nonzero.inds,]
sum(mycounts$log2fc >= 2)
[1] 1910
sum(meancounts$log2fc >= 2, na.rm = T)
[1] 1910
zero.inds <- which(meancounts[,1:2]==0,arr.ind=T)[,1]
mygenes <- meancounts[-zero.inds,]
sum(mygenes$log2fc >= 2)
[1] 314
How many genes are “down” regulated (at the -2 log2FC threshold)?
nonzero.inds <- rowSums(counts) != 0
mycounts <- meancounts[nonzero.inds,]
sum(mycounts$log2fc <= -2)
[1] 2330
sum(meancounts$log2fc <= -2, na.rm = T)
[1] 2330
zero.inds <- which(meancounts[,1:2]==0,arr.ind=T)[,1]
mygenes <- meancounts[-zero.inds,]
sum(mygenes$log2fc <= -2)
[1] 485
DESeq Analysis
Let’s do this with DESeq2 and put some stats behind these numbers
library(DESeq2)
Warning: package 'matrixStats' was built under R version 4.5.2
DESeq wants 3 things for analysis, countData, colData, and design.
dds <- DESeqDataSetFromMatrix(countData =counts,
colData = metadata,
design = ~dex)
converting counts to integer mode
Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
design formula are characters, converting to factors
The main function in the DESeq package to run analysis is called
DESeq().
dds <- DESeq(dds)
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
Get the results out of this DESeq object with the function results().
res <- results(dds)
head(res)
log2 fold change (MLE): dex treated vs control
Wald test p-value: dex treated vs control
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 747.194195 -0.3507030 0.168246 -2.084470 0.0371175
ENSG00000000005 0.000000 NA NA NA NA
ENSG00000000419 520.134160 0.2061078 0.101059 2.039475 0.0414026
ENSG00000000457 322.664844 0.0245269 0.145145 0.168982 0.8658106
ENSG00000000460 87.682625 -0.1471420 0.257007 -0.572521 0.5669691
ENSG00000000938 0.319167 -1.7322890 3.493601 -0.495846 0.6200029
padj
<numeric>
ENSG00000000003 0.163035
ENSG00000000005 NA
ENSG00000000419 0.176032
ENSG00000000457 0.961694
ENSG00000000460 0.815849
ENSG00000000938 NA
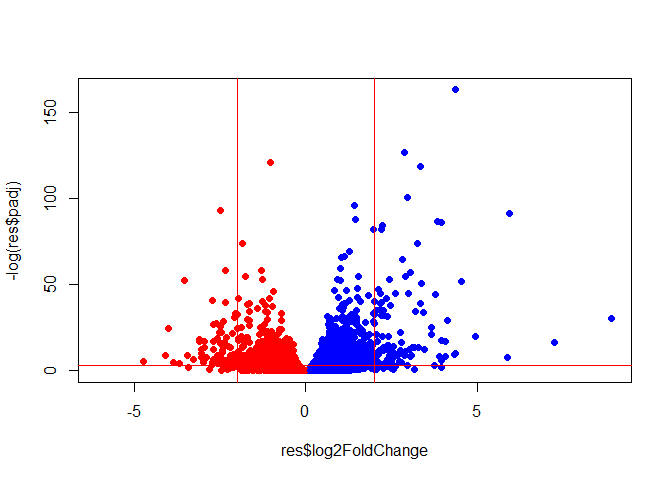
Volcano Plot
This is a plot of log2FC vs adjusted p-value
colors <- ifelse(res$log2FoldChange > 0, "blue", "red")
plot(res$log2FoldChange, -log(res$padj), col = colors, pch = 16)
abline(v = c(-2, 2), col = "red")
abline(h = -log(0.05), col = "red")
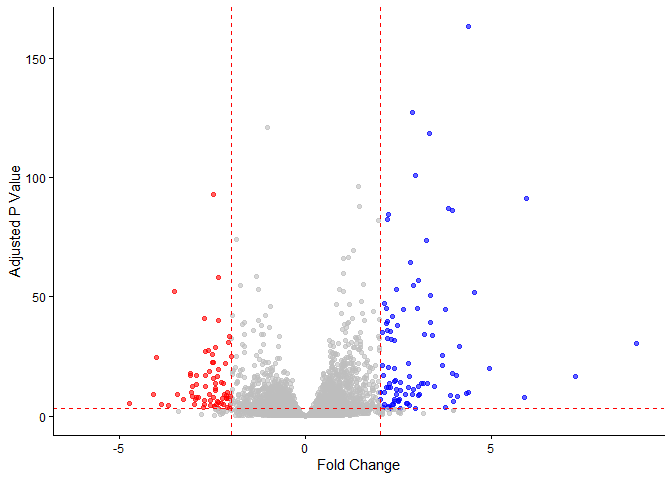
mycols <- rep("gray", nrow(res)) # Start with gray for all
mycols[res$log2FoldChange >= 2] <- "blue" # Positive large fold changes blue
mycols[res$log2FoldChange <= -2] <- "red" # Negative large fold changes red
mycols[res$padj >= 0.05] <- "gray" # Non-significant grey regardless
ggplot(res) +
aes(x = log2FoldChange, y = -log(padj)) +
geom_point(color = mycols, alpha = 0.6) +
theme_classic() +
geom_vline(xintercept = c(-2, 2), color = "red", linetype = "dashed") +
geom_hline(yintercept = -log(0.05), color = "red", linetype = "dashed") +
labs(x = "Fold Change", y = "Adjusted P Value")
Warning: Removed 23549 rows containing missing values or values outside the scale range
(`geom_point()`).
Save our results
write.csv(res,file="BGGN213_Class12Results.csv")
Pathway Analysis
library(pathview)
##############################################################################
Pathview is an open source software package distributed under GNU General
Public License version 3 (GPLv3). Details of GPLv3 is available at
http://www.gnu.org/licenses/gpl-3.0.html. Particullary, users are required to
formally cite the original Pathview paper (not just mention it) in publications
or products. For details, do citation("pathview") within R.
The pathview downloads and uses KEGG data. Non-academic uses may require a KEGG
license agreement (details at http://www.kegg.jp/kegg/legal.html).
##############################################################################
library(gage)
library(gageData)
data(kegg.sets.hs)
# Examine the first 2 pathways in this kegg set for humans
head(kegg.sets.hs, 2)
$`hsa00232 Caffeine metabolism`
[1] "10" "1544" "1548" "1549" "1553" "7498" "9"
$`hsa00983 Drug metabolism - other enzymes`
[1] "10" "1066" "10720" "10941" "151531" "1548" "1549" "1551"
[9] "1553" "1576" "1577" "1806" "1807" "1890" "221223" "2990"
[17] "3251" "3614" "3615" "3704" "51733" "54490" "54575" "54576"
[25] "54577" "54578" "54579" "54600" "54657" "54658" "54659" "54963"
[33] "574537" "64816" "7083" "7084" "7172" "7363" "7364" "7365"
[41] "7366" "7367" "7371" "7372" "7378" "7498" "79799" "83549"
[49] "8824" "8833" "9" "978"
foldchanges = res$log2FoldChange
names(foldchanges) = res$entrez
head(foldchanges)
[1] -0.35070302 NA 0.20610777 0.02452695 -0.14714205 -1.73228897
# Get the results
keggres = gage(foldchanges, gsets=kegg.sets.hs)
attributes(keggres)
$names
[1] "greater" "less" "stats"
# Look at the first three down (less) pathways
head(keggres$less, 3)
p.geomean stat.mean p.val q.val
hsa00232 Caffeine metabolism NA NaN NA NA
hsa00983 Drug metabolism - other enzymes NA NaN NA NA
hsa01100 Metabolic pathways NA NaN NA NA
set.size exp1
hsa00232 Caffeine metabolism 0 NA
hsa00983 Drug metabolism - other enzymes 0 NA
hsa01100 Metabolic pathways 0 NA
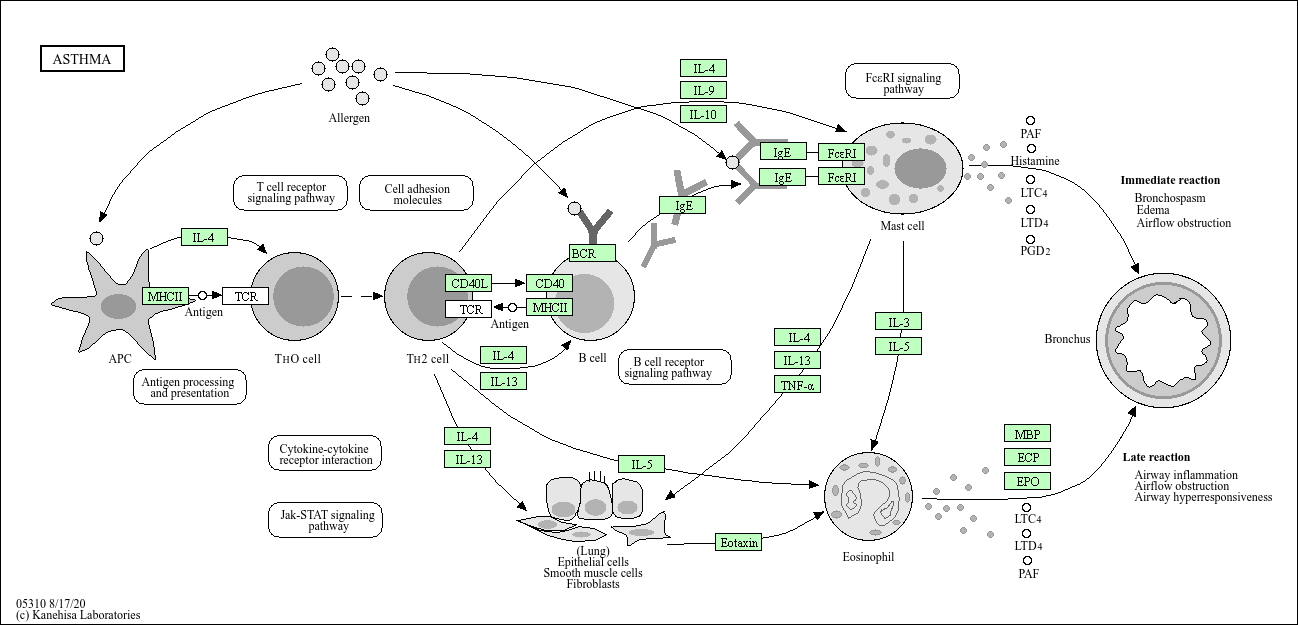
pathview(gene.data=foldchanges, pathway.id="hsa05310")
Warning: None of the genes or compounds mapped to the pathway!
Argument gene.idtype or cpd.idtype may be wrong.
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory C:/Users/kavan/Desktop/bggn213_f25_github/Class 12
Info: Writing image file hsa05310.pathview.png
 We will use the gage function from
Bioconductor.
We will use the gage function from
Bioconductor.
What gage wants as an inpiy is a simple named vector of importance (i.e. a vector with labeled fold changes)
library(gage)
library(gageData)
foldchanges <- res$log2FoldChange
names(foldchanges) <- res$entrezid
head(foldchanges)
[1] -0.35070302 NA 0.20610777 0.02452695 -0.14714205 -1.73228897
data(kegg.sets.hs)
keggres = gage(foldchanges, gsets=kegg.sets.hs)
head(keggres$less,5)
p.geomean stat.mean p.val q.val
hsa00232 Caffeine metabolism NA NaN NA NA
hsa00983 Drug metabolism - other enzymes NA NaN NA NA
hsa01100 Metabolic pathways NA NaN NA NA
hsa00230 Purine metabolism NA NaN NA NA
hsa05340 Primary immunodeficiency NA NaN NA NA
set.size exp1
hsa00232 Caffeine metabolism 0 NA
hsa00983 Drug metabolism - other enzymes 0 NA
hsa01100 Metabolic pathways 0 NA
hsa00230 Purine metabolism 0 NA
hsa05340 Primary immunodeficiency 0 NA
Let’s take a look at just one of these hsa05310
library(pathview)
pathview(gene.data=foldchanges, pathway.id="hsa05310")
Warning: None of the genes or compounds mapped to the pathway!
Argument gene.idtype or cpd.idtype may be wrong.
'select()' returned 1:1 mapping between keys and columns
Info: Working in directory C:/Users/kavan/Desktop/bggn213_f25_github/Class 12
Info: Writing image file hsa05310.pathview.png
Adḍ annotation data
We need to add gene symbols, gene names, and other database ids to make my results useful for future analyses.
head(res)
log2 fold change (MLE): dex treated vs control
Wald test p-value: dex treated vs control
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 747.194195 -0.3507030 0.168246 -2.084470 0.0371175
ENSG00000000005 0.000000 NA NA NA NA
ENSG00000000419 520.134160 0.2061078 0.101059 2.039475 0.0414026
ENSG00000000457 322.664844 0.0245269 0.145145 0.168982 0.8658106
ENSG00000000460 87.682625 -0.1471420 0.257007 -0.572521 0.5669691
ENSG00000000938 0.319167 -1.7322890 3.493601 -0.495846 0.6200029
padj
<numeric>
ENSG00000000003 0.163035
ENSG00000000005 NA
ENSG00000000419 0.176032
ENSG00000000457 0.961694
ENSG00000000460 0.815849
ENSG00000000938 NA
We have ENSEMBLE database IDs in our res object
head(rownames(res))
[1] "ENSG00000000003" "ENSG00000000005" "ENSG00000000419" "ENSG00000000457"
[5] "ENSG00000000460" "ENSG00000000938"
We can use the mapIDs() function from Bioconductor to help us:
library("AnnotationDbi")
library("org.Hs.eg.db")
Let’s see what database ID formats we can translate into
columns(org.Hs.eg.db)
[1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS"
[6] "ENTREZID" "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME"
[11] "GENETYPE" "GO" "GOALL" "IPI" "MAP"
[16] "OMIM" "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM"
[21] "PMID" "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG"
[26] "UNIPROT"
res$symbol <- mapIds(org.Hs.eg.db,
keys=row.names(res), # Our genenames
keytype="ENSEMBL", # The format of our genenames
column="SYMBOL", # The new format we want to add
multiVals="first")
'select()' returned 1:many mapping between keys and columns
head(res$symbol)
ENSG00000000003 ENSG00000000005 ENSG00000000419 ENSG00000000457 ENSG00000000460
"TSPAN6" "TNMD" "DPM1" "SCYL3" "FIRRM"
ENSG00000000938
"FGR"
Add GENENAME then ENTREZID
res$genename <- mapIds(org.Hs.eg.db,
keys=row.names(res), # Our genenames
keytype="ENSEMBL", # The format of our genenames
column="GENENAME", # The new format we want to add
multiVals="first")
'select()' returned 1:many mapping between keys and columns
res$entrezid <- mapIds(org.Hs.eg.db,
keys=row.names(res), # Our genenames
keytype="ENSEMBL", # The format of our genenames
column="ENTREZID", # The new format we want to add
multiVals="first")
'select()' returned 1:many mapping between keys and columns
head(res)
log2 fold change (MLE): dex treated vs control
Wald test p-value: dex treated vs control
DataFrame with 6 rows and 9 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 747.194195 -0.3507030 0.168246 -2.084470 0.0371175
ENSG00000000005 0.000000 NA NA NA NA
ENSG00000000419 520.134160 0.2061078 0.101059 2.039475 0.0414026
ENSG00000000457 322.664844 0.0245269 0.145145 0.168982 0.8658106
ENSG00000000460 87.682625 -0.1471420 0.257007 -0.572521 0.5669691
ENSG00000000938 0.319167 -1.7322890 3.493601 -0.495846 0.6200029
padj symbol genename entrezid
<numeric> <character> <character> <character>
ENSG00000000003 0.163035 TSPAN6 tetraspanin 6 7105
ENSG00000000005 NA TNMD tenomodulin 64102
ENSG00000000419 0.176032 DPM1 dolichyl-phosphate m.. 8813
ENSG00000000457 0.961694 SCYL3 SCY1 like pseudokina.. 57147
ENSG00000000460 0.815849 FIRRM FIGNL1 interacting r.. 55732
ENSG00000000938 NA FGR FGR proto-oncogene, .. 2268
Save my annotated results
write.csv(res,file="myresults_annotated.csv")